What Is DOCK?
DOCK addresses the problem of "docking" molecules to each other. In general, "docking" is the identification of the low-energy binding modes of a small molecule, or ligand, within the active site of a macromolecule, or receptor, whose structure is known. A compound that interacts strongly with, or binds, a receptor associated with a disease may inhibit its function and thus act as a drug. Solving the docking problem computationally requires an accurate representation of the molecular energetics as well as an efficient algorithm to search the potential binding modes.
Historically, the DOCK algorithm addressed rigid body docking using a geometric matching algorithm to superimpose the ligand onto a negative image of the binding pocket. Important features that improved the algorithm's ability to find the lowest-energy binding mode, including force-field based scoring, on-the-fly optimization, an improved matching algorithm for rigid body docking and an algorithm for flexible ligand docking, have been added over the years. For more information on past versions of DOCK, click here.
With the release of DOCK 6, we continue to improve the algorithm's ability to predict binding poses by adding new features like force-field scoring enhanced by solvation and receptor flexibility. For more information about the current release of DOCK, click here.
Why Use DOCK?
We and others have used DOCK for the following applications:
-
predict binding modes of small molecule-protein complexes
-
search databases of ligands for compounds that inhibit enzyme activity
-
search databases of ligands for compounds that bind a particular protein
-
search databases of ligands for compounds that bind nucleic acid targets
-
examine possible binding orientations of protein-protein and protein-DNA complexes
-
help guide synthetic efforts by examining small molecules that are computationally derivatized
-
many more...
Below are two examples of how DOCK can be used to predict the small molecule-target interactions. Variations on the same general procedure are used for all DOCKing applications.
Small Molecule Inhibitors of a Protein Drug Target
 This image graphically depicts a typical DOCK setup used toward the discovery of a potential disease inhibitor. The target (green) is an HIVgp41 protein, which mediates membrane fusion between the HIV virus and the host cell. During DOCKing, spheres (orange) are used to guide the placement of potential compounds (colored by atom type). These compounds are then scored using a grid (gray box) and ranked by their potential to bind to the target. (Courtesy of Dr. Robert Rizzo)
This image graphically depicts a typical DOCK setup used toward the discovery of a potential disease inhibitor. The target (green) is an HIVgp41 protein, which mediates membrane fusion between the HIV virus and the host cell. During DOCKing, spheres (orange) are used to guide the placement of potential compounds (colored by atom type). These compounds are then scored using a grid (gray box) and ranked by their potential to bind to the target. (Courtesy of Dr. Robert Rizzo)
Binding Pose Prediction for a Small Molecule in an RNA Active Site
1) Add protons, VDW parameters, and partial charges for both target and small molecule.

2) Calculate solvent accessible surface area of target (shown here colored by element).

3) Create negative image of surface features surrounding active site using spheres.

4) Calculate energy grid for target. Each grid point stores VDW score and charge for that area of space.
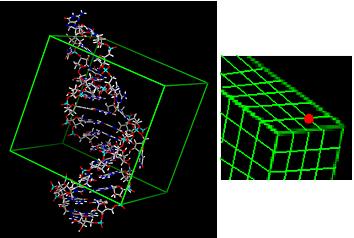
5) Match ligand atoms to sphere centers and score against grid.

6) Rank best scoring poses (top ten poses from DOCK run shown here).
